
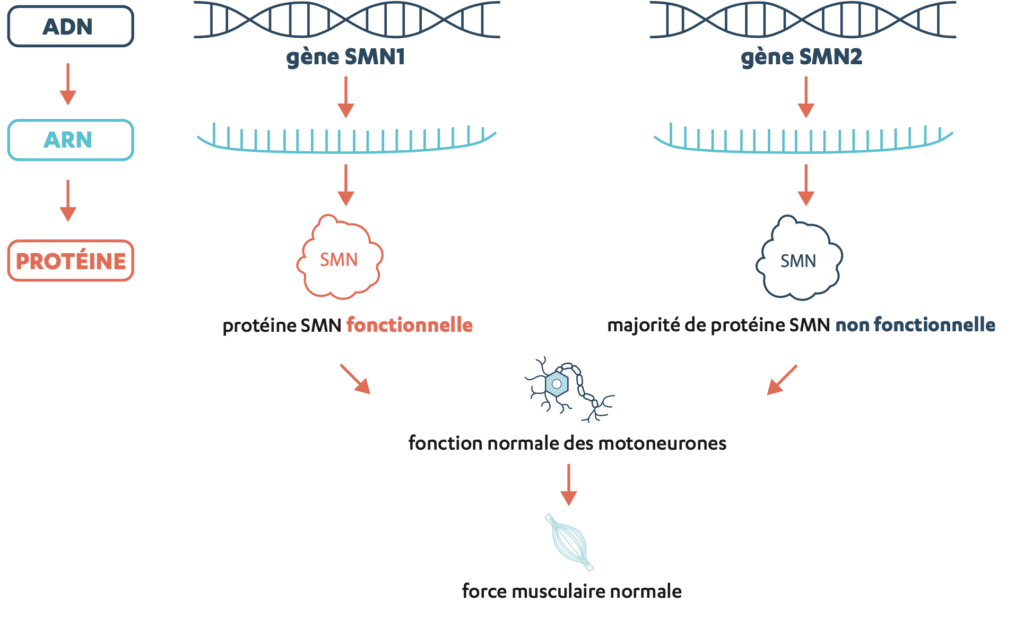
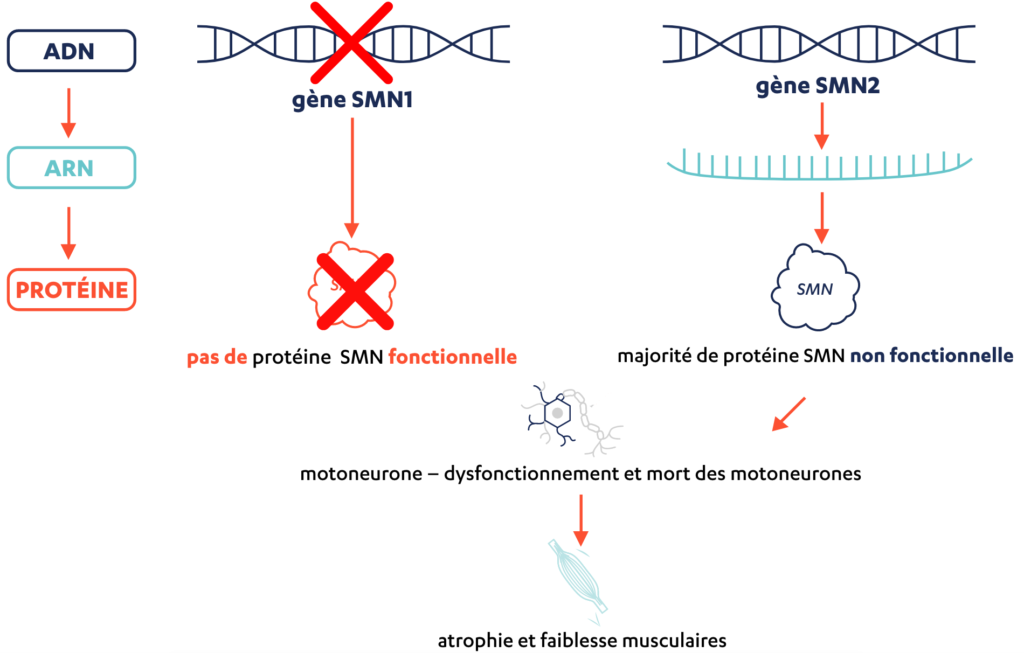









L’amyotrophie spinale (SMA) est une maladie rare, progressive et héréditaire qui provoque ou peut provoquer une faiblesse musculaire qui retentit souvent sur d’autres fonctions. Elle est due à l’altération (mutation) ou à l’absence (délétion) du gène SMN1 dans le patrimoine génétique (génome). Ce gène produit normalement le schéma de construction d’une protéine importante appelée la protéine de «survie du motoneurone» (SMN: Survival of Motor Neuron).
La protéine SMN est produite partout dans l’organisme et est nécessaire dans différentes cellules du corps. Elle est cependant particulièrement importante pour la fonctionnalité et la survie des motoneurones, des cellules nerveuses essentielles au bon fonctionnement des muscles. C’est pourquoi elle est également décrite comme facteur de protection des nerfs.
En plus du gène SMN1, la plupart des individus possèdent aussi le gène SMN2, qui fabrique également la protéine SMN. Si le gène SMN1 est manquant (délété) ou altéré (muté), le gène SMN2 peut «intervenir» (voir l’illustration en bas de la page 7). Cependant, le gène SMN2 produit principalement des protéines tronquées et incomplètes, et seulement une petite quantité de protéines SMN fonctionnelles.
ADN est l’acronyme de acide désoxyribonucléique. L’ADN porte le patrimoine génétique de tous les organismes vivants. Toutes les cellules du corps renferment de l’ADN dans leur noyau.
ARN est l’acronyme de acide ribonucléique. L’ARN joue un rôle clé dans la production des protéines, car il fournit des instructions pour leur construction.
En cas de déficit de SMN, les motoneurones «tombent malades» et finissent par mourir. Les muscles reçoivent de moins en moins d’impulsions nerveuses et ne peuvent plus fonctionner correctement; ils deviennent plus faibles et s’atrophient.
La SMA ne touche pas seulement les muscles squelettiques. Outre une mobilité de plus en plus réduite, les personnes atteintes de SMA peuvent présenter d’autres complications ou symptômes concomitants, tels que:
Problèmes de déglutition et d’alimentation
Faiblesse des muscles respiratoires et difficulté à expectorer, ce qui peut entraîner une susceptibilité accrue aux pneumonies et à d’autres problèmes respiratoires
De simples infections respiratoires peuvent rapidement conduire à des situations d’urgence
Scoliose (déviation de la colonne vertébrale) et déformations des os
Instabilité des hanches
Problèmes gastro-intestinaux: par exemple, reflux gastro-œsophagien, constipation due à un manque de motilité intestinale
Magen-Darm-Probleme: z.B. Reflux (Rückfluss des säurehaltigen Mageninhalts in die Speiseröhre), Verstopfung durch mangelnde Darmbewegung
Fatigue, fatigabilité accrue
Diminution de la densité osseuse, propension aux fractures
L’amyotrophie spinale diffère beaucoup d’une personne à l’autre. Comme chez les personnes atteintes de SMA le gène SMN1 ne peut pas fabriquer la protéine SMN, la quantité de protéine SMN dont elles disposent dépend en partie du nombre de copies du gène SMN2 qu’elles possèdent: plus les personnes atteintes de SMA ont de copies du gène SMN2, plus celui-ci pourra produire de protéine SMN fonctionnelle.
Bien qu’avec un nombre de copies plus élevé du gène SMN2, la maladie progresse généralement plus lentement ou est moins grave, il n’est pas recommandé de prédire la gravité de la SMA uniquement sur la base du nombre de copies du gène SMN2, car il peut y avoir des différences de gravité malgré un nombre égal de copies.
Les «limites» entre les différents types de SMA (habituellement décrits types 0 à 4) sont floues et il est impossible de prédire l’évolution de la maladie (pronostic) à partir de cette seule classification. Certaines personnes atteintes présentent également des symptômes qui se situent entre les types décrits. Par conséquent, il est plus judicieux d’adapter le traitement des personnes atteintes de SMA à leur état fonctionnel et à leurs symptômes. On distingue ainsi les personnes qui ne peuvent pas se tenir assises, celles qui peuvent se tenir assises et celles qui peuvent marcher.
La classification par type repose sur l’âge auquel les symptômes sont apparus pour la première fois et sur l’étape de développement moteur la plus avancée atteinte.
Les symptômes
apparaissent géné-
ralement avant l’âge
de 6 mois. La station
assise n’est jamais
possible.
Les symptômes apparaissent généralement entre 6 et 18 mois. La station assise est possible, la station debout n’est jamais possible.
Les symptômes apparaissent généralement après l’âge de 18 mois, mais ils peuvent apparaître tard dans l’enfance ou même au début de l’âge adulte. La station debout et la marche sont possibles mais risquent d’etre perdues avec l’évolution de la maladie.
Il existe entre outre deux formes moins courantes de SMA:
Maladies du Cancer