
Contenu
Traiter
Comment l’hémophilie est-elle traitée?
L’hémophilie étant un trouble de la coagulation, le traitement standard actuel consiste à administrer, en injection, le facteur de coagulation manquant. Mais les traitements qui accompagnent ces injections, comme la physiothérapie et les traitements contre la douleur, sont aussi des piliers thérapeutiques essentiels pour préserver la qualité de vie.
Vue d’ensemble des traitements de l’hémophilie
Un patient hémophile dispose aujourd’hui des options thérapeutiques suivantes:
| Principe thérapeutique | Administration | |
|---|---|---|
| Hémophilie A (absence ou déficit de facteur VIII) | Préparation à base de facteur de coagulation (remplacement du facteur VIII) | Voie intraveineuse (injection dans la veine du bras) |
| Anticorps bispécifique (assure la fonction du facteur VIII) | Voie sous-cutanée (injection directement sous la peau) | |
| Hémophilie B (absence ou déficit de facteur IX) | Préparation à base de facteur de coagulation (remplacement du facteur IX) | Voie intraveineuse (injection dans la veine du bras) |
Obtenez ici un aperçu des types d’injection.
Principe du traitement: remplacement du facteur de coagulation manquant
Depuis que l’on sait que l’hémophilie est due à un déficit ou à une absence totale d’un facteur de coagulation, ce qui empêche le sang de coaguler en cas de blessure, le traitement standard actuel intervient à ce niveau: on remplace le facteur de coagulation correspondant – facteur VIII en cas d’hémophilie A et facteur IX en cas d’hémophilie B – par une préparation à base de ce facteur ou par un anticorps.
Lorsque les traitements par un facteur de substitution ont fait leur apparition dans les années 1950, le facteur manquant était extrait du sang d’un donneur humain avec d’autres protéines. Ce mélange de protéines obtenu selon la méthode de fractionnement de Cohn était administré en injection intraveineuse aux patients hémophiles en présence d’une hémorragie aiguë. Il permettait à la cascade de la coagulation de se terminer et de stopper ainsi de manière fiable l’hémorragie.
Quelques années plus tard, les scientifiques sont parvenus à extraire séparément le facteur adapté à chaque type d’hémophilie à partir du plasma humain. Cette avancée a permis d’administrer aux patients hémophiles uniquement le facteur qui leur manquait (préparation à base de facteur plasmatique), à savoir le facteur VIII ou le facteur IX. Depuis maintenant plus de 10 ans, on trouve sur le marché des préparations à base de facteurs plasmatiques et d’autres à base de facteurs recombinants. La plupart des patients s’administrent ces concentrés à la maison (autotraitement à domicile) et n’ont plus besoin d’aller régulièrement à l’hôpital.
Recherche et développement dans le domaine de l’hémophilie
Le médicament à base du facteur manquant était initialement produit à partir du sang de donneurs. Au début, le concentré de facteur apportait donc au patient non seulement le facteur qui lui manquait, mais pouvait aussi lui transmettre des agents pathogènes du donneur. De nombreux hémophiles ont ainsi été infectés par les virus de l’hépatite B et C. Dans les années 1980, la transmission du VIH par des médicaments à base de facteurs de coagulation a été particulièrement dramatique.
On a ensuite cherché des méthodes pour rendre ces médicaments plus sûrs et on utilise depuis lors des procédés spécifiques pour éliminer, ou inactiver, les agents pathogènes (virus et bactéries) dans les médicaments produits à partir de plasma humain. D’autres travaux de recherche ont permis la fabrication du facteur manquant par voie biotechnologique, sans recourir à du sang humain. Des médicaments fabriqués par des procédés exclusivement biotechnologiques, médicaments dits recombinants, sont disponibles en Suisse depuis 2004.
Une préparation pour administration sous-cutanée destinée au traitement prophylactique de l’hémophilie A sévère est disponible depuis 2019. Un anticorps bispécifique, fabriqué par le biais d’un procédé biotechnologique, assure dans le corps la fonction du facteur VIII manquant ou présent en quantité insuffisante, ce qui accélère la coagulation. Cette préparation est injectée directement sous la peau (voie sous-cutanée).
Prophylaxie ou traitement à la demande: tout dépend de la sévérité de la maladie
Les facteurs manquants sont non seulement utilisés pour le traitement aigu, mais aussi – selon la sévérité de l’hémophilie – à titre préventif (prophylaxie). Chez les patients présentant une hémophilie modérée à sévère, la prophylaxie permet de réduire les saignements spontanés et de minimiser les lésions articulaires sur le long terme.
De nombreux patients peuvent ainsi mener aujourd’hui une vie quasi normale. Une règle doit toutefois être respectée: les préparations à base de facteurs de coagulation doivent être administrées à intervalles réguliers. Les facteurs de coagulation subissent en effet une dégradation dans le foie. La vitesse de cette dégradation– également appelée «demi-vie» – varie d’un individu à l’autre et en fonction de la préparation. Pour déterminer la fréquence à laquelle le médicament doit être administré, on calcule la demi-vie du facteur dans le sang.
La demi-vie correspond au délai jusqu’à ce que la quantité de facteur présente dans le sang soit réduite de moitié. Si la demi-vie est de douze heures, il ne reste plus que la moitié de la quantité initiale de facteur douze heures après son administration. Au bout des douze heures suivantes, il ne reste plus que la moitié de la moitié et ainsi de suite. Au bout d’un certain temps, la quantité de facteur VIII présente dans le sang sera donc très faible. Il faudra alors injecter une nouvelle dose de facteur pour que la coagulation continue à fonctionner. Par conséquent, plus la demi-vie est longue, moins souvent le patient doit s’administrer le médicament. La notion de demi-vie est aussi clairement expliquée dans la vidéo (en allemand) de l’Association Suisse des Hémophiles.
Les inhibiteurs compliquent le traitement
Une complication majeure dans la substitution d’un facteur de coagulation sanguine vient de l’émergence d’un inhibiteur qui réduit considérablement l’efficacité du facteur substitué.
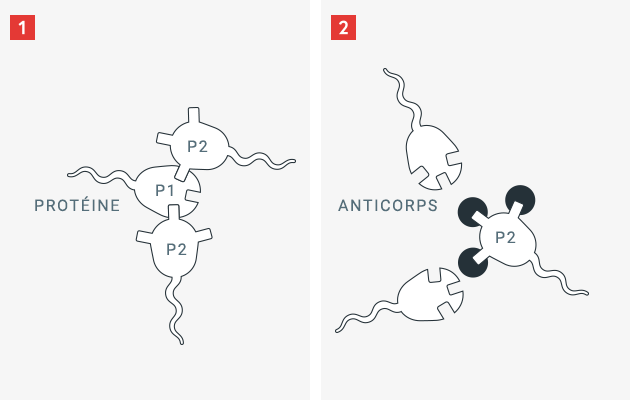
Le système immunitaire humain est conçu pour protéger notamment contre l’intrusion d’agents pathogènes dans l’organisme, tels que les bactéries, les virus ou les champignons. Pour se défendre de ces parasites omniprésents, le système immunitaire a acquis l’aptitude à faire la différence entre les propriétés de structures étrangères et les structures et protéines propres à l’organisme. Dès que le système immunitaire détecte une protéine étrangère, les cellules immunitaires de l’organisme produisent, après une série d’étapes intermédiaires, des substances de défense, appelées anticorps.
Les anticorps sont des protéines capables de se fixer avec une grande spécificité sur des structures en surface définies, permettant ainsi à l’organisme de neutraliser une protéine étrangère et de la dégrader plus facilement. Ce processus n’a pas lieu avec des protéines propres à l’organisme humain, ce dernier – et son système immunitaire dans le cadre de son développement et de sa maturation – ayant appris à différencier entre « étranger » et « endogène ». Un patient atteint d’un déficit héréditaire de facteur de coagulation sanguine ne possède pas cette protéine de coagulation spécifique, ou seulement sous une forme modifiée (et donc moins active), c’est pourquoi son système immunitaire n’a pas appris à identifier comme « endogène » cette protéine de coagulation normalement présente. Si l’on administre au patient la protéine de coagulation nécessaire (par ex. le facteur VIII), il est possible que le système immunitaire l’identifie comme « étrangère » et produise des anticorps spécifiques pour la combattre.
Dans le cas de l’hémophilie, ces anticorps portent aussi le nom d’inhibiteurs. Ces anticorps spécifiques peuvent bloquer ou affaiblir l’activité de la protéine de coagulation administrée ou encore raccourcir sa durée de vie. Le médicament administré ne protège donc plus le patient contre les hémorragies.
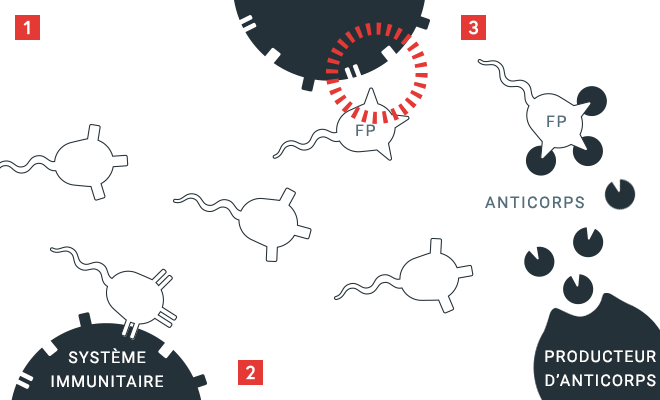
Principe de la formation d’anticorps
Le système immunitaire contrôle les protéines aussi selon des caractéristiques « endogènes » qui lui sont familières. S’il identifie des protéines comme « étrangères » (FP), il produit des anticorps qui se fixent sur ces caractéristiques inconnues. La formation d’inhibiteurs génère la production d’anticorps qui s’attaquent au facteur VIII injecté, lequel perd donc toute efficacité.
Facteurs de risque favorisant la formation d’inhibiteurs
Les troubles de la coagulation ne comportent pas tous un risque égal de production d’inhibiteurs dans le cadre d’un traitement par substitution (apport du facteur manquant). Le risque de la formation d’inhibiteurs est le plus élevé au cours des 50 premières administrations du produit de substitution, puis baisse jusqu’à la 150e injection pour se stabiliser à un niveau faible et ne plus varier.
La production d’inhibiteurs dépend de facteurs de risque congénitaux, donc impossibles à influencer comme par ex. l’origine ethnique ou l’existence d’un parent porteur d’inhibiteurs, ou encore de facteurs de risque acquis, ces derniers pouvant être minimisés et faisant partie de la planification du traitement entre le médecin et le patient. Si le patient présente des inhibiteurs et saigne, il faut continuer de le traiter afin que les saignements ne lui laissent pas de séquelles.
Approches thérapeutiques
En cas de trouble de la coagulation sanguine, il existe aujourd’hui plusieurs traitements possibles qui permettent d’activer la production de fibrine et, par suite, de rétablir une coagulation sanguine correcte.
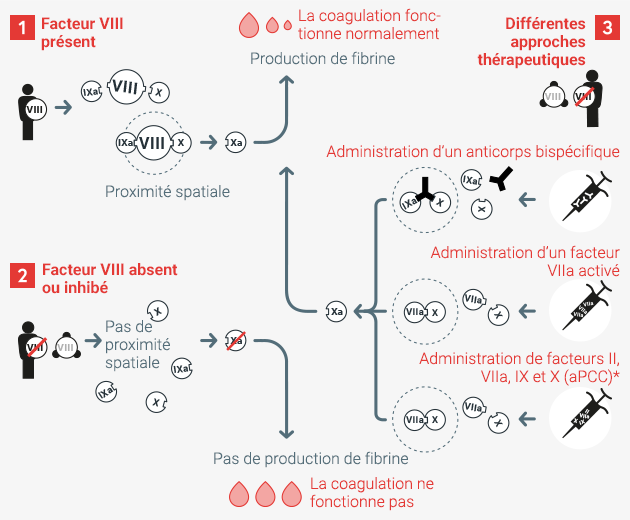
Approches thérapeutiques en cas de dérèglement de la coagulation sanguine *aPPC = concentré de facteurs du complexe prothrombique activé
Cas 1
Lorsque la coagulation sanguine est normale, le facteur VIII assure, en proximité spatiale étroite avec les facteurs IX et X, la production efficace de facteur Xa. Ce dernier est indispensable pour la production de fibrine, responsable de la coagulation du sang.
Cas 2
En l’absence du facteur VIII, ou s’il est inhibé par la fixation d’anticorps, la proximité spatiale avec les facteurs IXa, VIIIa et X fait défaut. La production de facteur Xa est perturbée et la coagulation sanguine enrayée.
Cas 3
L’administration d’un anticorps bispécifique établit une proximité spatiale étroite entre l’agent actif et les facteurs IXa et X, sans avoir besoin du facteur VIII. Il est également possible que l’administration du facteur VIIa activé, ou en association avec l’aPPC (concentré de facteurs du complexe prothrombique activé), entraîne la production du facteur VIII et, ainsi, garantisse une bonne coagulation sanguine.
Dans les trois cas, la production de facteur Xa fonctionne correctement, et, de ce fait, la coagulation sanguine également.
Il est possible de « bannir » un inhibiteur hors du corps : le système immunitaire apprend à tolérer une protéine de coagulation administrée suite à un traitement, appelé « induction de la tolérance immune ». Cette forme de traitement peut durer des mois ou des années et est très contraignante pour le patient. Chez certains patients présentant une forme particulière d’inhibiteurs, on supprime le système immunitaire par un traitement médicamenteux afin de neutraliser la production d’inhibiteurs
- Schweizerische Hämophilie-Gesellschaft www.shg.ch
- World Federation of Hemophilia www.wfh.org
- Selpers www.selfers.com
- Deutsche Hämophiliegesellschaft www.dhg.de
- Schmerz Nachrichten OÄ Dr. Waltraud Stromer: Hämophilie und Schmerztherapie (pdf)
- Uniklinikum Saarland Dr. med. Susan Halimeh: Die Bedeutung der Physiotherapie in der modernen Hämoph…